Rocket Introduction
Pre-requisites
This guide is designed for people just starting with High-Performance Computing at Newcastle University.
Please email me if you notice anything that is out of date, as I want to keep this current whilst I work here.
The guide assumes that you have been taught some Linux before, and have got a login on Aidan.
If the terms Aidan, unix.ncl.ac.uk, or SSH are alien to you, book yourself on a Linux Intro course (the Bioinformatics Support Unit run many) and ask about the following link: https://services.ncl.ac.uk/itservice/technical-services/unix-time-sharing/
Someone in your lab who is an admin for Rocket groups will need to have added you onto their group, to give you access to Rocket itself. If you haven’t been given access, talk to your PI.
Getting onto Rocket
Let’s start right at the beginning, with logging into Rocket. Rocket, like Aidan, is a Linux server that is accessed using SSH. If you are on campus, you should be able to SSH directly into Rocket using your preferred method. If not, Rocket is within the main Uni firewall, making it invisible unless you go through the Aidan Gateway server. In that case, SSH into Aidan, then SSH through into Rocket.
The following parameters should get you in.
| Parameter | Value |
|---|---|
| Username | Usual university username |
| Hostname | rocket.hpc.ncl.ac.uk |
| Port | 22 |
| Password | Usual university password |
What is Rocket?
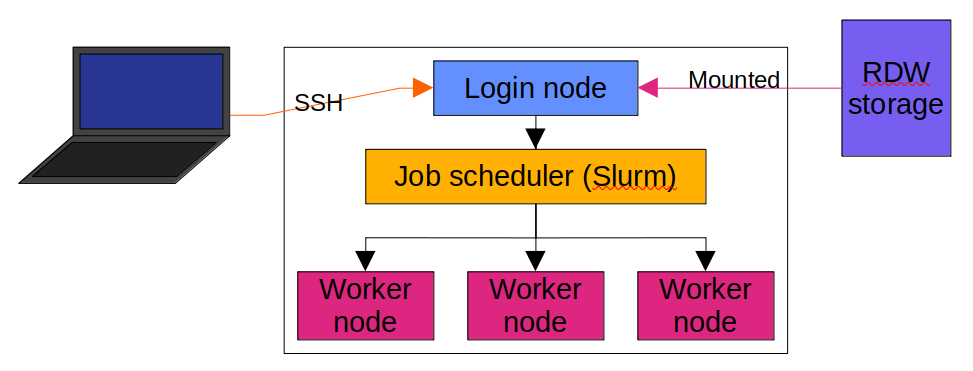
Rocket is a ‘cluster’ of computers, all working together to process a lot of data at once. These computers, called nodes, are connected to each other (as above) and are able to transfer data to one another. Rocket is made up of a couple of login nodes, which you interact with directly, a job scheduler, and some worker nodes which are not normally accessible by the user.
The login nodes don’t have good CPUs, or a lot of RAM, so are not designed for processing data in a big way. They are designed to be accessible to you, so you can sit on them and manipulate them directly, and external storage devices like the Research Data Warehouse (RDW) drive are accessible from there.
The worker nodes are separate computers which are somewhat hidden from the user, in that they are not normally able to be accessed by you. They do, however, have all of the large compute resources that are available to the cluster: CPUs with lots of cores, lots of RAM, etc.. More info about this is available at: https://services.ncl.ac.uk/itservice/research/hpc/hardware/
The job scheduler is a piece of software which handles the load in the cluster, as there are many more people who want to run analyses on the cluster than there are resources that the cluster can provide. The job scheduler puts people’s jobs in a queue, and prioritises these jobs when allocating them to the worker nodes, depending on what resources each job needs, and what’s available on the different nodes.
Basically, how does interacting with Rocket work?
You connect your PC/laptop/whatever to one of the login nodes of Rocket via SSH, and write scripts that are able to run on Rocket. You submit these scripts to the job scheduler along with inputs and outputs of the script, as a job. The job scheduler will put that job in a queue to be run. When there is space on one or more worker nodes for the job to run, the scheduler will allocate the whole node, or just a part of it, to your job, and the job will be run on that part.
Your first job on Rocket
This is a set of instructions that will give you a chance to download and locate data within Rocket, and to run simple jobs on it.
Setup
Before running a job, you will need some input data to run a job on. Let’s grab a FASTA file from the NCBI database, and put it onto Rocket.
- First, SSH into Rocket.
- You should land in your home, but worker nodes CAN NOT see your home directory.
You will need to go to your scratch space, which is available to worker nodes, but is not backed up.
cd /nobackup/yourUsername - Make a directory for this tutorial called SlurmTutorial and go into it:
mkdir SlurmTutorial cd SlurmTutorial - Download a file from the web with a link that points to that file.
In this case, the command points to the NCBI database, and to a file containing the sequence for the pUC18 bacterial plasmid.
The
-Oargument defines the output filename aspUC18.fasta.wget 'https://www.ncbi.nlm.nih.gov/sviewer/viewer.cgi?tool=portal&save=file&log$=seqview&db=nuccore&report=fasta&id=209211&filename=sequence.fasta' -O pUC18.fasta
Now, you have a FASTA file in a place that can be processed by the cluster.
Write your script
- Open up Nano in your terminal with
nano - Copy the following text into nano:
#!/bin/bash -l ################################################################################ # Slurm env setup # # Set number of cores #SBATCH -c 1 # Set RAM per core #SBATCH --mem-per-cpu=100M # Set mail preferences (NONE, BEGIN, END, FAIL, REQUEUE, ALL) #SBATCH --mail-type=NONE # Set queue in which to submit: defq bigmem short power #SBATCH -p short # Set wall clock time #SBATCH -t 0-0:10:00 # # ################################################################################ md5sum pUC18.fastaThis script looks a lot like it has a lot of comments and doesn’t do a lot, but that’s not true. Lines starting with
#are treated like comments by Bash, and aren’t read, but Slurm looks at lines starting with#SBATCHand takes them to be setup options for your job. In this case, the options request 1 CPU core and 100MB of RAM, for 10 minutes in the short queue. More about queues later. Once the job is set up, then the worker node will run themd5sumcommand on a file calledpUC18.fastathat is in the same directory that you are in when you submit the job. - Save the script in Nano by pressing the Control key and the X key at the same time (usually abbreviated to
Ctrl + X.- Press
Ywhen it asks you to save the buffer. - Enter the filename:
md5sum_pUC18.slurmat the prompt for the filename.
- Press
- Now, whilst you are in the directory:
/nobackup/yourUsername/SlurmTutorial, it’s time to submit the job!sbatch --job-name=MyFirstSlurmJob md5sum_pUC18.slurmThe
sbatchcommand tells Bash to submit the job to Slurm. Now, instead of defining the job name in the script with a#SBATCHline, you can define the job name after thesbatchcommand as above. Add the name of the Slurm script that you made earlier, and that is it! - Have a look at the output once it’s done.
If you
lsin this directory, there may be a file that looks something likeslurm-29454702.out. If not, your job may still be in the queue/running. In that case, check the queue with this exact command (don’t substitute anything):squeue -u $USEROnce there is nothing in that output, then the file should be generated. Have a look in the output with:
less slurm-29454702.outsubstituting my filename for yours. To get out of
less, press theQkey.
Storage space and the code of conduct
Please read the Rocket Code of Conduct before you start doing anything on the cluster. This is important.
Remember: The storage space that is visible to the worker nodes is in /nobackup/yourUsername/ and your home directory IS NOT VISIBLE!
More detail about Slurm on Rocket - queues and other Slurm commands
Slurm have a longer cheatsheet for all of the commands that are available here
The script in the Your first job on Rocket section was submitted to the short queue.
There are a few other queues that you can submit jobs to, which are detailed here, under Running jobs: https://services.ncl.ac.uk/itservice/research/hpc/using-gettingstarted/
Modules and installing software
Many people use Rocket at once from all over the university, and everyone needs different software.
For example, a mathematician is unlikely to need a tool for processing genetics data.
To keep things clean, not every available piece of software is loaded at once, and tools that are installed are kept in modules that are ready to be called into action.
To list all available software held in modules, you could use the module avail command, but that lists everything in a massive list, which is not very useful.
If you have the name of a tool and you want to see if it is installed on Rocket, use:
module avail toolName
where toolName is the name of the tool.
If a tool is listed as available, then load the tool with module load like so:
- Search for a tool called Samtools. This usually takes a while the first time after you login.
module avail samtools - Load the desired version:
module load SAMtools/1.16.1-GCC-11.3.0Most of the time, you just want the latest version, but sometimes, you need a specific version, so multiple are installed.
Once you have tested your modules on the login node, make sure to add a module load command to your Slurm script so that the worker node knows to load the same modules before running your analyses!
Detailed info about installin software on here is incomplete. Ask someone to show you how. Sufficed to say, you don’t have root access on Rocket, so you will need to bear that in mind when you follow installation guides online.
Where to get more information
Normally, SAgE run courses on Intro to HPC and Intro to Linux, which are available here: https://workshops.ncl.ac.uk/public/sage/
NUIT have their main documentation here: https://services.ncl.ac.uk/itservice/research/hpc/
Sample job scripts from NUIT are available here: https://services.ncl.ac.uk/itservice/research/hpc/jobscripts/ Most members of the Ryan Lab, including me, use scripts of the form in the basic example above to run CPU and/or RAM intensive jobs.
The FAQ for working with Rocket is here: https://services.ncl.ac.uk/itservice/research/hpc/using-faq/